
Dr. Daniel Balazs and Eliad Peretz will attend the 69th Lindau Nobel Laureate Meeting taking place from 30 June to 05 July 2019 in Lindau, Germany (https://www.lindau-nobel.org/).
Category Archives: Uncategorized
Curtis’s paper on isomerization of magic dots appears in Science.
For years, scientists have been trying to discover the size at which solid materials could change their internal structure in a single, swift step, like molecules do during isomerization. This unanswered question has been the missing link in scientists’ quest to map and understand the crossover from molecular isomerization, such as those that make eyesight possible, to bulk phase transitions, like the transition of graphite into diamonds. If understood, these processes could be useful for applications such as energy harvesting or quantum computing. In their recent paper published in Science (DOI: 10.1126/science.aau9464), Professors Tobias Hanrath and Richard Robinson finally reveal that a “magic size cluster” bridges this divide between how matter rearranges in the small scale of molecular isomerization and in large, solid bulk matter phase transitions.
Here is a link to the Cornell Chronicle article
http://news.cornell.edu/stories/2019/02/tiny-particles-can-switch-back-and-forth-between-phases
and the Science article,
http://science.sciencemag.org/content/363/6428/731
Ben’s latest paper on nanowire growth online
Ben’s work on the mechanistic details of growing nanowires on heated copper surfaces is published in Chemistry of Materials. The work discusses the thermodynamics of the seed formation, and shows a beautiful, simple method to study the process in situ. Read the paper HERE.
Congratulations!
Happy holidays and a productive new year!
The Hanrath Energy Lab wishes you happy holidays and a new year rich in results, grants and obtained skills and knowledge!

CANFI team wins commercialization and prototyping award
The CANFI (continuous additive nanomanufacturing at fluid interfaces) team won a commercialization and scale-up grant from the college of engineering.
Welcome new group members
Rileigh Casebolt and Jesus Miguel Lopez Baltazar join the group. Rileigh will be working on electrochemical CO2 reduction and Jesus will work on programmable assembly (in collaboration with Prof. Alabi)
Dr. Ben Richards! successfully defends his thesis. Congratulations Ben!
Congratulations, Dr. Richards! for successfully defending your PhD thesis. Pictures from the defense and abstract of the presentation are below.

post-defense champagne.

graduation ceremony

Q&A session

‘the audience’
Bulk-nucleated vapor-solid-solid Silicon and Germanium Nanowires: Synthesis, Scaling, and Applications
Benjamin T. Richards
Cornell University, 2018
Nanowires are essential building blocks for many next-generation devices. This includes applications in solar cells, field effect transistors, thermoelectric generators, chemical sensors, and electrochemical cells. One application of intense promise is using silicon and germanium nanowires as anodes materials in lithium ion batteries, as these materials form stable compounds with superior lithium capacity that increases the anode charge capacity compared to current Li-ion batteries by a factor of 4 – 10. Unlike the bulk material, nanowires have been demonstrated to withstand the ~400% volume dilation due to the intercalation of lithium. To bring the acclaimed potential of nanowires to fruition, challenges in production, scaling, and electrochemical performance must be resolved. Conventional nanowire synthesis methods produce small yields of nanowires that require many processes (i.e., slurry mixing, coating, baking) to introduce into batteries. To meet the growing energy storage demands for personal electronics and electric vehicles, an improved method is required to produce nanowires.
In this work, I will introduce a growth mechanism that is simple, robust, and adaptable to high throughput processing of nucleating and growing nanowires from bulk metal films, known as bulk nucleated vapor-solid-solid (BN-VSS). This method prepares nanowires that are epitaxially connected to the metal substrate, allowing direct integration into batteries. I identify the kinetic conditions required to grow nanowires, the processes that occur during nanowire growth, and copper as a promising growth substrate material. Then, I will present a kinetic model of germanium nanowire growth from copper films using diphenylgermane as a precursor. This model was developed by performing rapid syntheses of nanowires using an inductive heating apparatus followed by ex-situ characterization of these films. Finally, I introduce a diagnostic technique of measuring the electrical resistance of the growth substrate to monitor the solid-state transformation that proceeds to germanium nanowire growth and demonstrate how this method can be used to inform kinetic conditions in high-throughput roll-to-roll reactors.
‘The pulse of electrochemical CO2 reduction’ Kevin Kimura’s paper on the cover of ChemSusChem
Kevin’s recent paper on electrochemical CO2 reduction showed that pulsing the applied potential provides a rich parameter space of previously under appreciated ‘knobs’ to tailor the selectivity of reduced products. More information can be found in the Full Paper by Kimura et al. (https://onlinelibrary.wiley.com/doi/abs/10.1002/cssc.201801130).

Electrochemical CO2 reduction reaction (CO2RR) has garnered strong interest as a promising pathway to convert CO2 emissions into higher value chemicals including fuels and hydrocarbon feedstocks. In particular, copper has been shown to produce a range of useful hydrocarbons at room temperature, which could be powered renewably. The main challenges involved with CO2RR is the low selectivity to one product, competition with the hydrogen evolution reaction (HER) and the overall high overpotentials required. Applying the electrochemical potential in a time-programmed pulse (instead of a constant potential) has interesting implications on both fundamental and applied aspects of CO2RR. From a fundamental perspective, the timing of the square wave pulsed potential provides insights into the coupled dynamics of mass transport and surface reactions. From an applied perspective, pulsed potentials significantly mitigate electrode fouling in electrolytic cells. Intrigued by the prospects of pulsed potentials applied to CO2RR we sought out to understand the underlying mechanism responsible for pulse dependent product selectivity.
We found that the application of a pulsed potential allows us to substantially suppress the HER while shifting selectivity to CH4 and CO. We attribute the improved CO2RR selectivity to a re-arrangement of surface hydrogen coverage during the pulsing. We also establish that the size and geometry of the electrode matters; when using a small copper electrode, HER was suppressed to less than 5% and methane or CO could be selectivity produced even at fairly low potentials. To decouple the interplay of surface reactions and mass transport to and from the electrode we performed rotating disk electrode experiments and compared the results to an analytical model.
On a fundamental level, our findings provide new insights into the timescales of competing pathways in CO2RR. From an applied perspective, our results present an opportunity for the product selectivity to be tuned by adjusting the temporal profile of the electrochemical potential. We anticipate our findings may help others to further understand the CO2RR pathways and improve performance for other electrocatalysts.
Doug and Curtis publish their paper on the relationship of mesophase behavior and magic cluster stabilization
Congratulations to Doug and Curtis for publishing their paper in JACS.
Nevers, Douglas R., Curtis B. Williamson, Benjamin H. Savitzky, Ido Hadar, Uri Banin, Lena F. Kourkoutis, Tobias Hanrath, and Richard D. Robinson. “Mesophase Formation Stabilizes High-purity Magic-sized Clusters.” Journal of the American Chemical Society (2018). (https://pubs.acs.org/doi/abs/10.1021/jacs.7b12175)
 Magic-sized clusters (MSCs), which are identical in size and resistant to conventional nanoparticle (NP) growth processes, represent an ideal synthetic product to overcome the complications resulting from the size dispersion (at least 3%) inherent to conventional NP synthesis method. However, fundamental reaction pathways toward their nucleation and stabilization still present many outstanding challenges. Here, we show that our high concentration synthesis accentuates fundamental surfactant mesophase behavior that stabilize and enable isolation of high-purity cadmium sulfide MSCs.
Magic-sized clusters (MSCs), which are identical in size and resistant to conventional nanoparticle (NP) growth processes, represent an ideal synthetic product to overcome the complications resulting from the size dispersion (at least 3%) inherent to conventional NP synthesis method. However, fundamental reaction pathways toward their nucleation and stabilization still present many outstanding challenges. Here, we show that our high concentration synthesis accentuates fundamental surfactant mesophase behavior that stabilize and enable isolation of high-purity cadmium sulfide MSCs.
In this paper we address a fundamental question for MSCs: how does the precursor concentration direct the synthetic pathway between NPs and MSCs? Building upon our recently developed synthesis methods we demonstrate that a crossover in behavior is achieved by tuning concentration: ultra-high concentration synthesis promotes a well-defined reaction pathway to produce high purity MSCs (>99.9%) and forms a mesophase assembly that kinetically arrests the MSCs from NP growth. Using in-situ X-ray scattering, we find that high-purity MSC formation is accompanied by the production of a large hexagonal mesophase assembly (>100 nm grain size), containing thousands of discrete MSCs. At intermediate concentrations the mesophase assembly is less stable, resulting in NP growth at the expense of the large MSC assemblies. Collectively, these findings present an alternative result from the conventional mantra that the stability of MSCs derives from the precise arrangement of the inorganic structures (i.e., closed-shell atomic packing); we demonstrate that disordered, yet distinct, clusters can also be stabilized by self-forming fibrous mesophase assemblies. In this light we evaluate previous MSC studies and suggest that they may have been, unknowingly, observing mesophase formation or surfactant phase behavior coupled with their MSC formation. Overall, we find that the high concentration approach intensifies and showcases inherent concentration-dependent surfactant phase behavior that is not accessible in conventional (i.e., dilute) conditions.
We propose that our high concentration synthesis of MSCs provides not only a robust method to synthesize, stabilize, and study identical NP products, but also demonstrates an underappreciated stabilizing role of surfactants in NP synthesis.
Kevin’s review paper on Entropic, enthalpic and kinetic aspects of interfacial nanocrystal superlattice assembly and attachment now in press
Kevin’s review paper titled “Entropic, enthalpic and kinetic aspects of interfacial nanocrystal superlattice assembly and attachment” is now available: http://pubs.acs.org/doi/10.1021/acs.chemmater.7b04223
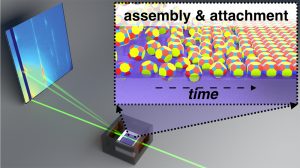
The directed assembly of nanoscale building blocks into complex superstructures is of widespread scientific and technological interest. Scientists and engineers have been intrigued by the prospects of tailoring self-assembly processes to create materials whose properties and function can be tuned through the interaction between constituent particles. In particular, Recent reports of epitaxially connected CQD superlattices with long-range atomic coherence have generated significant interest as a platform for novel, quasi 2D ‘designer materials’.
The coupled thermodynamic and kinetic principles governing the interfacial nanoparticle self-assembly and directed attachment present a rich, albeit complex scientific problem. In the enclosed manuscript, we describe the interesting interplay of entropic and enthalpic driving forces and the kinetic aspects of interfacial self-assembly and attachment. We present in-situ grazing incidence X-ray scattering measurements and emerging insights into the complex choreography of interfacial transport processes involved in the formation of highly ordered epitaxially connected nanocrystal solids. New understanding emerging from in-situ measurements provides process control and design principles for the selective formation of specific superlattice polymorphs. We discuss outstanding challenges that must be resolved to translate know-how from controlled assembly and attachment in the laboratory to scalable integration for emerging technological applications.