
Congratulations to Kevin, Rileigh and the rest of the team for publishing their work in ACS Catalysis (10.1021/acscatal.0c02630)
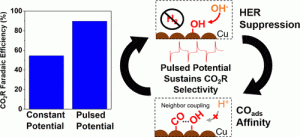
Electrochemical CO2 reduction (CO2R) on copper has garnered strong interest as a promising pathway to convert CO2 emissions into higher value chemicals including fuels and hydrocarbon feedstocks. We previously reported (Kimura et al. ChemSusChem (2018)) that the application of a pulsed potential during CO2R improved selectivity toward converting CO2 to higher order products, suppressed undesirable H2 production, and maintained electrode stability. The working hypothesis which emerged from the initial study was that the pulsing-dependent product selectivity was related to dynamic changes in species adsorbed to the electrode surface.
In this study we set out to establish deeper fundamental insights into the mechanism by applying in-situ techniques. We used in-situ X-ray adsorption spectroscopy to identify and monitor the bulk Cu valence state remained reduced during the pulsing, but found surface hydroxides (OHads) were adsorbing during the anodic pulsed potential. We combined these spectroscopic insights with the recent findings by Iijima et al. (ACS Catalysis (2019)), in which in-situ attenuated total reflection surface-enhanced infrared absorption spectroscopy was used to demonstrate the effect of surface hydroxides promoting CO adsorption and preventing Cu deactivation. We concluded the pulsed mechanism favors CO2 reduction and increases stability due to two effects: 1) proton desorption/displacement by OHads during the anodic potential, which suppresses H2 production and 2) the accumulation of OHads, promoting COads and preventing Cu deactivation.